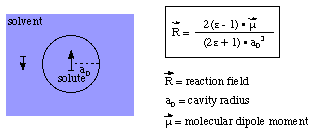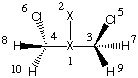| medium | dielectric
constant | vib1 | vib2 | vib3 | vib4 | vib5 | vib6
|
|---|
| vapor | 1.00 | 1167 | 1249 | 1500 | 1746 | 2782 | 2843
| | acetonitrile | 36.64 | - | 1247 | 1503 | 1723 | 2797 | 2876
| | | | | | | |
| | difference | - | - | -2 | +3 | -23 | +15 | +33
|
| |
 |
These trends can be reproduced semi-quantitatively using the Onsager reaction field model
in combination with Hartree-Fock theory or hybrid density function methods such as Becke3LYP
and medium sized basis sets such s 6-31G(d) or 6-31+G(d).
One more interesting application of the Onsager reaction field model concerns the optimization
of molecular structures that are not even stationary points in the gas phase. This is particularly
usefull for strongly polarized (charge separtated) structures and the SN2 substitution
reaction of primary amines with alkyl halides (Menshutkin reaction) will be used as an example here.
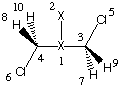
If we use ammonia as a simple amine and methyl chloride as the reaction partner, we should assume
that the SN2 process leads to the formation of a complex of the methylammonium cation
and the chloride anion. This latter structure is, however, energetically so unfavorable in the
gas phase that neither a transition state nor the product complex can be optimized.
#P Becke3LYP/6-31+G(d) freq
b3lyp/6-31+G(d) opt reactant complex NH3 + MeCl
0 1
X1
C2 1 1.0
Cl3 2 r3 1 90.0
N4 2 r4 1 a4 3 180.0
H5 2 r5 3 a5 1 180.0
H6 2 r6 5 a6 3 d6
H7 2 r6 5 a6 3 -d6
H8 4 r8 2 a8 1 0.0
H9 4 r9 2 a9 8 d9
H10 4 r9 2 a9 8 -d9
r3=1.81282958
r4=3.39606276
r5=1.08870123
r6=1.08876628
r8=1.01866066
r9=1.01851992
a4=90.5440056
a5=108.63033934
a6=110.29980273
a8=111.86983212
a9=111.56759592
d6=-118.94994045
d9=-120.07441969
| |
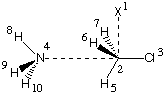 |
However, starting from the gas phase reactant complex and using the Onsager
reaction field for aqueous solution (dielectric constant = 78.39), it is possible
to calculate a complete reaction profile containing the reactant and product complexes
as well as the SN2 transition state. The actual shape of the solution phase
PES will, of course, depend significantly on the chosen cavity radius.
last changes: 01.04.2008, AS
questions & comments to: axel.schulz@uni-rostock.de
|