Force Field Methods
In force field or molecular mechanics (MM) methods the molecules are treated as mechanically connected systems of atoms. In contrast to quantum mechanical methods, electrons are not treated explicitly but together with the nuclei as effective atoms. The energy expressions used in MM methods are chosen such that the system is well described at typical bonding distances only. The description of reactions or transformations in which bond breaking and making occurs is therefore not possible with most approaches. A given force field usually contains a number of different interaction terms describing different types of strain possible in a molecular system. The following is a list of components that are typically considered:
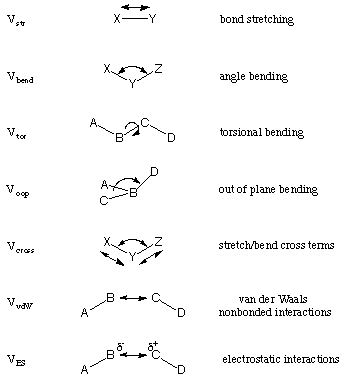
The overall (strain) energy of the system is then given as the sum of all these terms over all occurences in the system:
EMM = Vstr + Vbend + Vtor + Voop + Vcross + VvdW + VES
Van der Waals and electrostatic parameters are usually not calculated for atoms that are at a distance of one or two bonds in a given molecule. The closest interacting centers for these types of interactions are separated by at least three bonds and relative to each other in 1-4 position. These types of interactions are therefore often referred to as 1,4-interactions.
While the smallest interacting unit in all atom (AA) force fields are the single atoms of the system, groups of atoms are used as interacting units in united atom (UA) force fields. In particular for the modeling of large systems (such as proteins or polymers) it is quite sufficient to treat, for example, CH3 - groups as one effective interaction center that is slightly larger than a single carbon atom. Even in all atom force fields, however, different parameters are used to describe centers of different bonding characteristics. Typically there will be different parameters for sp3-, sp2-, and sp-type carbon atoms and additional ones for carbon atoms in carbonyl groups or aromatic systems.
Typical energy expressions for some of the enery terms are as follows:
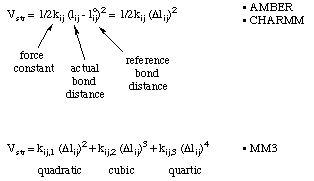
Most force fields use a simple quadratic (Hook's law) energy expression with only one term. In order to evaluate this energy expression for a given structure, the reference bond distance (for an unstrained bond) and a force constant describing the ease of deformation are needed. These expressions are used for example by the AMBER or CHARMM force fields. Somewhat more elaborate bond stretching functions containing cubic and quartic terms are used by the MM3 hydrocarbon force field.
A similar functional form is used for the angle bending deformation:
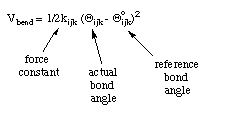
Again an unstrained reference bond angle is assumed, whose deformation is possible with force constant kijk.
The functions describing the torsional potential energies are different from the previous terms as they must be periodic with respect to rotation by 360 degrees:
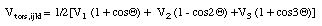
The use of all three potential parameters V1, V2, and V3 is not necessary in all cases.
There are several possibilities for the description of van der Waals interactions between non-bonded atoms, the Lennard-Jones 6-12 potential being the most commonly used:
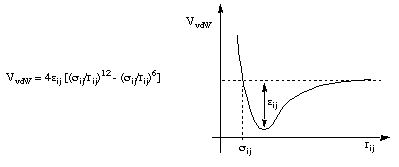
Here the interaction between two non-bonded atoms are described as slightly attractive at longer distances, but strongly repulsive at closer distances. Two potential parameters are needed for each non-bonded interaction type.
The energy expression for the electrostatic interactions usually follows a simple Coulomb law, which is always attractive for partial charges of opposite sign and always repulsive for partial charges of equal sign:
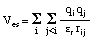
The parameters needed for evaluation of this expression are the partial charges q for the interacting centers i and j as well as an effective dielectric constant. As calculation of non-bonded interactions becomes quite time consuming for extended systems, most programs use a cutoff value of between 9 and 16 Angstroms, beyond which the interactions are tailed off to zero over an additional short distance.
In evaluating, deriving and comparing force field parameters for any of these potential energy functions it is important to remember, that all of these parameters are only valid within a given force field, that is, a set of other parameters and potential energy functions. A single force field parameter has not to be meaningful by itself! And there are many different parameters and functions that yield the same final result (strain energy)!