Internal Coordinates
Geometry optimization in internal coordinates is achieved using the
opt=Z-Matrix
keyword. In this case the coordinate system used in the input file is also that used for geometry optimization. The success of the geometry optimization (number of optimization cycles, final result of the optimization) therefore depends critically on the coordinates used in the input file. The optimization in internal coordinates (as given in Z-Matrix format) is particularly useful in the following three cases:
1) Highly symmetric systems
The benfits of using internal coordinates for highly symmetric systems can nicely be demonstrated using ammonia NH3 as an example. Ammonia is a slightly pyramidalized amine of C3v symmetry. A first attempt for a Z-Matrix would be as follows:

The three N-H bonds have been defined using different variables for bond distances and bond angles, but the initial values have been chosen such that all three N-H bonds are of equal length, implying a certain degree of symmetry. When this input is used, Gaussian only recognizes Cs symmetry since the three H-N-H angles are not identical. This type of geometry definition also holds the danger of leading to changes in the point group during geometry optimizations. The latter are indicated by the program as:
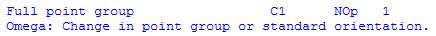
This error typically occurs when optimization starts at a highly symmetric structure which looses some of its symmetry properties on optimization due to an incorrect definition of Z-Matrix variables. In the rare case that the symmetry of the system should not be used, one can actually force the program to ignore symmetry completely using the
NoSymm
keyword. In most cases, however, the available symmetry properties will be used to reduce the computational effort (in part considerably). Returning to ammonia as an example, a better set of internal coordinates is:

reducing the number of variables from six to three. Using this input, Gaussian still only recognizes Cs symmetry, but smoothly optimizes the structure of ammonia in five steps to tight conversion criteria. Optimal use of the symmetry properties of ammonia can be made using one dummy atom designated with the symbol "X" located on the principal C3 axis:
<

In this case the number of variables can be reduced to two since the definition of the relative position of the hydrogen atoms uses the intrisic properties of the threefold principal axis. The dihedral angles of 120.0 degrees must remain constant during the geometry optimization and these values are therefore not defined through variables, but as numerical constants in the Z-Matrix. Using the above Z-Matrix, the full C3v symmetry of ammonia is recognized by Gaussian, but optimization is consequently performed in the Cs point group due to the lack of support for threefold symmetry axes. At the HF/6-31G(d) level of theory, the geometry is optimized to tight convergence criteria in five cycles with optimized parameters of r3=1.002522 and a3=111.6755, the HF/6-31G(d) total energy for this structure being -56.1843563au.
One complication sometimes arising from the use of dummy atoms is that one looses sight of the actual number of degrees of freedom. A nonlinear, asymmetric molecular system containing N atoms has 3N-6 degrees of freedom (=independent structural parameters). For linear, asymmetric systems this number is increased to 3N-5. Using ammonia again as an example, we should have 3*4-6 = 6 independent structural parameters if symmetry is not accounted for. We have seen above that this number can be reduced to 2 on full consideration of its C3v symmetry. How many degrees of freedom remain after dummy atoms have been used in Z-Matrix construction is, however, rather difficult to see in some cases. One can therefore instruct Gaussian to check whether the structural variables used in a given Z-Matrix are all linearly independent (that is, describe different structural degrees of freedom) and overall don't exceed the correct number in a given point group symmetry. This is done by using
FOpt instead of Opt
as the keyword. One example, that demonstrates a problematic situation, is the following:

Here the position of dummy atom X1 is indirectly optimized through its use for the definition of hydrogen atom H4. Running this input with the FOpt keyword gives the following error:
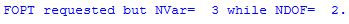
In most cases, the optimization of the position of dummy atoms leads to larger numbers of geometry optimization cycles and in some cases to incomplete optimizations.
2) Systems containing rings
The construction of ring systems is a particular challenge due to the fact that one of the ring bonds cannot be specified in a Z-Matrix. We will use oxirane (C2H4O, ethylene oxide) here as an example to demonstrate the situation. Oxirane is C2v symmetric. A Z-Matrix reflecting this symmetry is as follows:
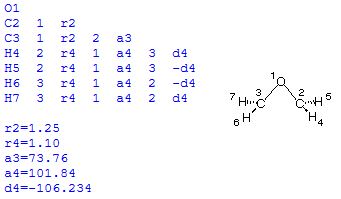
With this definition, the distance between C2 and C3 is not defined explicitly as a variable, but indirectly through variable a3 describing the bond angle C3-O1-C2. Small variations in this angle will, unfortunately, lead to quite large changes in the C2-C3 distance. As a consequence, convergence of the optimization is less efficient and takes 8 steps to complete. Alternatively, the geometry can also be defined through inclusion of one dummy atom located on the principal C2 axis:
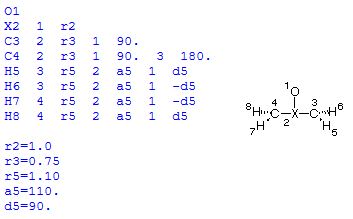
In this Z-Matrix all bond distances inside the ring are defined through a distance variable, even though these variables do clearly not describe what is typically considered a "bond distance". Optimization is more efficient and is complete within 7 steps. The difference between these two approaches increases with system size and complexity.
3) Linear Systems
A suitable example for a molecule containing a linear arrangement of atoms is acetonitrile (CH3CN), one of the more common solvents used in organic and biological chemistry. This system belongs to the C3v point group and contains a linear C-C-N arrangement. A first attempt at a Z-Matrix is:

The use of this Z-Matrix leads, however, to the following error already during the first step of the geometry optimization:

What is located on "card 6" (=line 6 of the Z-Matrix) is the definition of the position of atom 6. At the heart of the problem is the definition of dihedral angles in general. By definition, dihedral angle D(1/2/3/4) is the angle between two planes, the first being described by atoms 1/2/3 and the second one described by atoms 2/3/4. As soon as atoms 1/2/3 or 2/3/4 fall on one line, the definition of one of the planes has vanished and the dihedral angle is undefined. One could attempt to "solve" the situation by bending the nitrile group a little bit out of linearity, introducing one additional variable:

Since support for the principal C3 axis is lacking anyway, one could as well start with a Cs symmetric starting structure, but introduce threefold symmetry through the backdoor using equal C-H bond lengths and H-C-C bond angles. This time the program runs for a few optimization cycles until the C-C-N bond angle (variable a6) approaches very closely to 180.0 degrees. Gaussian then stops either with the error message cited above or (more appropriately) with:

It thus appears that this strategy doesn't represent a good solution to the problem of large bond angles either. Dividing the critical bond angle in two parts through introduction of a dummy atom, however, solves the problem:

In this case the optimization is smoothly performed to tight convergence criteria within 5 steps at the HF/6-31G(d) level of theory with final values of r2=1.467835, r3=1.082125, r7=1.134723, a3=109.8344 and a final total energy of -131.9275339au. Similar situations exist in many other compounds containing C-C or C-N triple bonds as well as transition metal complexes containing CO-ligands.
In conclusion, optimization in internal coordinates is particularly helpful for highly symmetric systems, for which the Z-Matrices can be constructed easily through the appropriate use of dummy atoms. Bond distances should always be positive, bond angles between 0.0 and 180.0 degrees, and dihedral angles between 0.0 and 360.0 degrees.